肠道菌群作为人体内复杂的微生态系统,主要由9个门的细菌和1种古菌组成,可参与合成及分泌维持器官和免疫系统正常功能的基本物质,
因此肠道菌群也被认为是人类的另一个内分泌系统。近年来被发现在代谢疾病的发病机制中扮演关键角色。
本文将带您探索肠道菌群与肥胖、糖尿病等代谢疾病的密切关系,了解科学家们如何揭开这一奥秘,并介绍基于这些发现的前沿治疗方法。
首先,肠道菌群是如何被发现与代谢相关的呢?相关研究发现肥胖人群的肠道菌群组成与瘦人明显不同,特别是两类主要细菌——厚壁菌门和拟杆菌门的比例存在显著差异。
2006年,Jeffrey Gordon教授团队进行了一项开创性实验:将肥胖小鼠的肠道菌群移植到无菌小鼠体内,结果接受移植的小鼠即使进食相同量的食物,体重也显著增加,这直接证明了肠道菌群对体重的因果性影响。
那么肠道菌群是如何影响肥胖的呢?肠道菌群影响肥胖的分子机制,涵盖能量提取、胆汁酸代谢、免疫调节、肠-脑轴调控等关键途径。
一、能量代谢调控
1、短链脂肪酸(SCFAs)的双重作用
短链脂肪酸(SCFAs,如乙酸、丙酸、丁酸)是肠道菌群发酵膳食纤维的主要代谢产物,通过以下机制影响肥胖:
(1)能量供应:乙酸和丙酸被宿主吸收,作为能量底物(占每日能量需求的5-10%),促进肝脏糖异生和脂肪合成。
(2)受体介导的信号调控:
GPR41/43激活:SCFAs通过G蛋白偶联受体(GPR41/43)抑制脂肪分解,促进瘦素分泌,调节食欲。
HDAC抑制:丁酸作为组蛋白去乙酰化酶(HDAC)抑制剂,上调PPARγ表达,促进脂肪细胞分化。
2、胆汁酸代谢重塑
肠道菌群通过7α-脱羟基作用将初级胆汁酸(CA、CDCA)转化为次级胆汁酸(DCA、LCA),影响宿主代谢:
FXR信号通路:次级胆汁酸激活法尼醇X受体(FXR),抑制肝脏脂肪生成。
TGR5-GLP-1轴:胆汁酸通过TGR5受体促进L细胞分泌GLP-1,增强胰岛素敏感性。
二、免疫调节代谢
肠道菌群失衡引发炎症导致肥胖机制链条:
高脂饮食/菌群失调 → 肠道屏障损伤 → LPS(脂多糖)入血 → TLR4/NF-κB激活 → 慢性炎症 → 胰岛素抵抗+脂肪堆积
详细步骤解析:
1、肠道屏障破坏:
高脂饮食减少紧密连接蛋白(如occludin、ZO-1),使肠道上皮细胞间隙增大。如大肠杆菌等革兰氏阴性菌增殖,释放更多LPS(脂多糖)。
2、LPS进入循环:
正常情况下,LPS被肠道黏液层阻挡。但当黏液层变薄,LPS穿过屏障进入门静脉,引发”代谢性内毒素血症”(血浆LPS水平升高2-3倍)。
3、TLR4/NF-κB通路激活:
LPS与免疫细胞表面的TLR4受体结合,触发NF-κB信号,导致:
(1)促炎细胞因子释放:TNF-α、IL-6水平升高
(2)胰岛素信号抑制:炎症因子使胰岛素受体底物(IRS)丝氨酸磷酸化,阻断正常信号传导
(3)脂肪分解受阻:炎症促进脂肪细胞脂解酶(如ATGL)活性下降,脂肪堆积加剧
当体内发生慢性炎症后:慢性炎症→胰岛素抵抗→血糖升高→胰腺β细胞代偿性分泌更多胰岛素→高胰岛素血症促进脂肪合成(尤其内脏脂肪)。
三、肠-脑轴调控
1.食欲调控
(1)迷走神经信号:SCFAs(如丙酸)通过迷走神经抑制下丘脑NPY/AgRP神经元,减少摄食食欲。
(2)5-HT(血清素):肠道5-HT(占全身90%):主要由肠道嗜铬细胞和菌群产生,肠道菌群通过色氨酸代谢影响5-HT合成,高5-HT水平抑制棕色脂肪产热,减少能量消耗,增加脂肪的储存而引发肥胖。
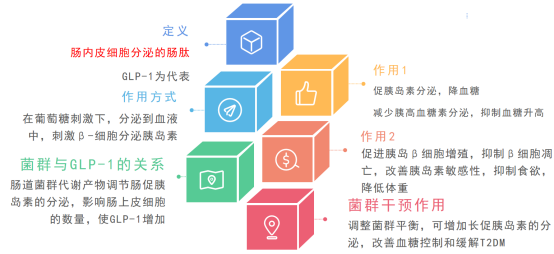
2.GLP-1与PYY分泌
肠道L细胞在接触:短链脂肪酸(如丁酸)、胆汁酸(特别是次级胆汁酸)时会分泌
肠道内分泌细胞:SCFAs和胆汁酸刺激L细胞分泌GLP-1和PYY,
其中GLP-1能:1)延缓胃排空(食物停留更久);2)直接作用于大脑产生饱足感;3)刺激胰岛素分泌(但不像药物会引发低血糖)。而PYY与GLP-1协同工作:1)高蛋白饮食后显著升高;2)抑制胃肠蠕动;3)减少对食物气味的敏感度。
肠道菌群与2型糖尿病(T2DM)的关联已被大量研究证实。
2型糖尿病患者肠道菌群功能的改变可能会引发肠道微生态的中度失衡。体内产丁酸细菌丰度的下降会引起硫酸盐还原及氧化应激抵抗的生物聚集,导致炎症反应。
研究显示,2型糖尿病患者体内菌群与健康人群存在明显差异。
2型糖尿病患者体内梭状芽孢杆菌和厚壁菌的比例显著低于健康人群,
而伴随机体糖耐量的降低,拟杆菌与大肠埃希菌的比值有所升高,证实了在2型糖尿病的发生发展过程中,肠道菌群存着一定的失衡。
菌群失衡可通过能量代谢紊乱、肠屏障损伤、慢性炎症等多条途径促进糖尿病发生。
一、能量代谢紊乱
短链脂肪酸(SCFAs)减少 → 胰岛素抵抗产丁酸菌(如Faecalibacterium prausnitzii、Roseburia intestinalis)、产丙酸菌(如Bacteroides)等这些菌群发酵膳食纤维生成丁酸、丙酸等SCFAs。
丁酸可增强肠道屏障,减少炎症,促进GLP-1分泌(改善胰岛素敏感性);丙酸抑制肝脏糖异生,降低血糖。
而T2DM患者产SCFAs菌减少,导致GLP-1分泌不足,进而导致一以下情况:
1. 胰岛素分泌受损(β细胞功能下降)
GLP-1减少→胰岛素分泌延迟;餐后血糖峰值更高(>10 mmol/L);长期高血糖进一步损伤β细胞(”糖毒性”)
2. 胰高血糖素抑制失效(α细胞失控)
GLP-1不足→胰高血糖素异常升高→肝糖输出增加→空腹血糖升高。
3. 胃排空加快 → 餐后血糖飙升
T2DM患者胃排空加速(1-2小时),导致葡萄糖吸收过快,加重血糖波动。
4. 中枢性食欲调控失调
GLP-1减少→大脑持续接收”饥饿信号”→进食量增加→肥胖风险↑。
除了GLP-1下降,SCFAs缺乏还会直接加剧胰岛素抵抗:
1. 脂肪组织炎症增加
丁酸抑制HDAC(组蛋白去乙酰化酶)→减少促炎因子(TNF-α、IL-6)→改善脂肪细胞胰岛素敏感性。
SCFAs不足→脂肪组织巨噬细胞向M1型(促炎)极化→胰岛素信号受阻。
2. 肝脏糖异生失控
丙酸通过FFAR3受体抑制肝脏糖异生关键酶(PEPCK、G6Pase)。
SCFAs减少→肝脏过度生成葡萄糖→空腹血糖升高。
3. 肌肉葡萄糖摄取减少
丁酸促进肌肉细胞AMPK活化→增加GLUT4转位→提升葡萄糖摄取。
T2DM患者肌肉葡萄糖摄取率下降40%,与SCFAs缺乏相关。
二、肠屏障损伤
肠道菌群失衡导致的肠道屏障破坏(俗称”肠漏”)是2型糖尿病发生的关键环节。
首先菌群失衡是如何破坏屏障的?
菌群失调导致:
促炎细胞因子(TNF-α)↑ → 下调ZO-1蛋白表达;丁酸(修复紧密连接)↓
屏障被破坏后糖尿病发生链条:
1. 内毒素(LPS)入血
革兰氏阴性菌(如大肠杆菌)的LPS通过破损屏障进入门静脉
糖尿病患者血清LPS水平比正常人高2-3倍
2. 免疫系统过激反应
LPS结合免疫细胞TLR4受体
激活NF-κB通路 → TNF-α、IL-6等炎症因子爆发
临床数据:T2DM患者CRP水平平均升高3mg/L
3. 胰岛素信号被”干扰”
TNF-α使胰岛素受体底物(IRS-1)发生丝氨酸磷酸化
阻断正常的酪氨酸磷酸化信号传导
结果:葡萄糖转运蛋白GLUT4无法转位 → 细胞”拒绝”吸收血糖
4. 胰腺β细胞代偿衰竭
长期高血糖迫使β细胞超负荷工作
炎症环境加速β细胞凋亡
研究显示:确诊糖尿病时β细胞功能已丧失50%
三、慢性炎症
肠漏(Leaky Gut)→ 慢性炎症 → 胰岛素抵抗
黏液降解菌(如Akkermansia muciniphila,保护屏障)、产LPS菌(如Escherichia coli、Enterobacter,破坏屏障)等肠道菌群在其中发挥重要作用。
高脂/高糖饮食→A. muciniphila减少→黏液层变薄→肠道通透性↑
LPS(内毒素)进入血液→激活免疫系统(TLR4/NF-κB通路)→慢性低度炎症
炎症因子(TNF-α、IL-6)干扰胰岛素信号→胰岛素抵抗